
THE DISEASE
APERT SYNDROME
|
In the case of Apert’s, the spheno-occipital and spheno- ethmoidal synchondroses and the fronto-ethmoidal suture fuse early, resulting in a severely shortened posterior/anterior cranial base with a resultant hypoplastic midface.[7] The most readily observed malocclusions are a severe maxillary anterior open bite and a severely crowded and retrusive maxillary causing impactions, severe crowding of developing teeths, delayed eruption, thick gingiva, and in some cases supernumerary teeth [4] or congenitally missing teeth. Furthermore, sleep apnea and dry mouth are resulting conditions. Complex syndactyly of the phalanges in the hand and foot arise causing webbing due to fusion.[1,7]
|

Image revealing how sutures have per-maturely fused cause deformation and and inlets of bones that fill the gap caused
|
CRANIOSYNOSTOSES
|
Cranyiosynostosis syndromes, such as Apert syndrome, share phenotypes that include; premature
closing of cranial sutures, limb
abnormalities and in some cases central nervous system malformations.
Most craniosynostosis syndromes result from missense mutations in FGFR2. [18]
Premature suture fusion results in abnormalities of the head. These changes of head shape can be associated with increased intracranial pressure that may result in permanent brain inury. In addition to the risks of brain inury, craniosynostoses are associated with: alteration of craniofacial growth leading to mid-facial hypoplasia, abnormalities in dental alignment and orbital deformation. [19] The combination of craniosynostisis and its associated facial malformations leads to significant morbidity and mortality. Patients often require skills of large interdisciplinary medical and surgical team and often undergo multiple reconstructive surgeries to try and correct some of the issues. [19] These conditions place a great burden on the pateints their families and the health care system. Since the genetic etiology of human craniosynostoses is only partially understood, what researches have discerned is that Noggin, a protein, is required for embryonic neural tube development as well as for skeleton patterning. In addition noggin has been shown to be expressed postnatal in the sutre of mesenchyme, through not fusing cranial sutures regulated by FGF-based signaling. [16] As Noggin misexpression prevents cranial suture fusion in vitro and in vivo it has been suggested that FGFR mediated craniosynostoses may be a result of inappropriate down regulation of Noggin expression. Increased FGF signaling might lead to suture fusion by suppressing Noggin production in the dura mater and osteoblasts of normally patent canial sutures. [16] |
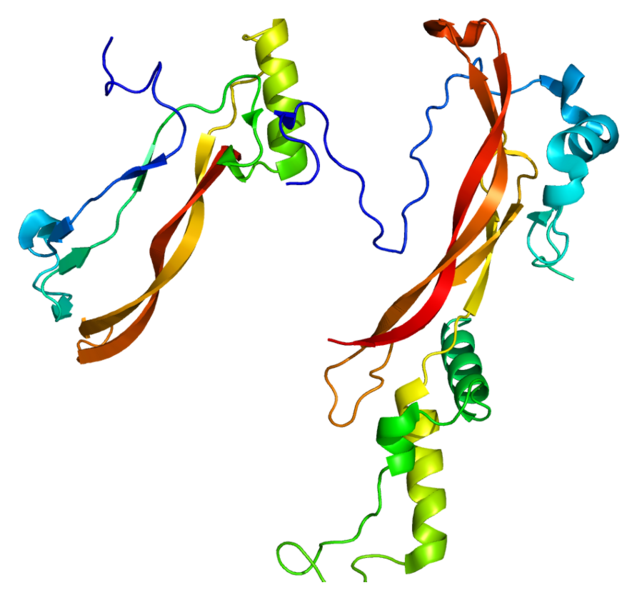
The protein Noggin, involved in bone development, joint development and neural tube fusion
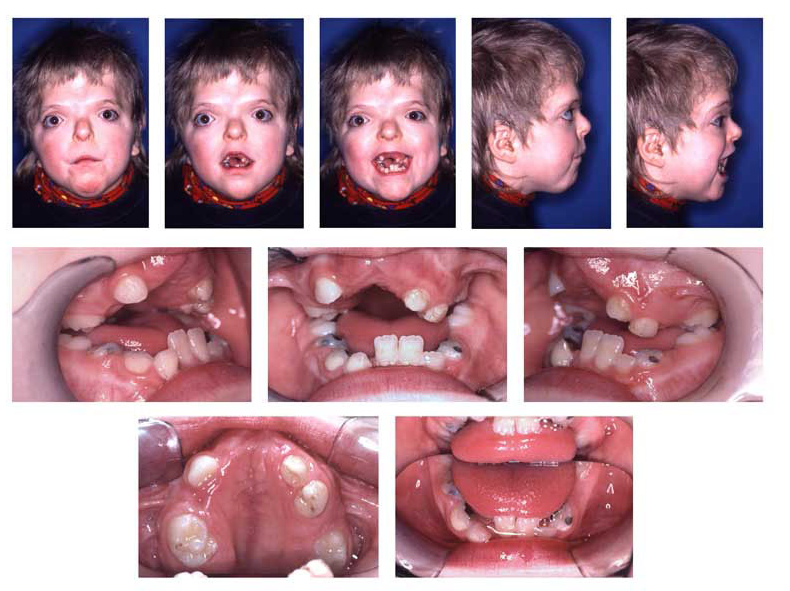
Photographic example of Apert syndrome with this infant displaying the characteristic symptoms
|
